Prion protein N-linked glycosylation: review and assessment of therapeutic potential
This post will review what is known about PrP glycosylation and its role (if any) in prion diseases, and will examine whether glycosylation represents a potential therapeutic target for treating these diseases.
what is glycosylation?
Glycosylation is the attachment of sugar chains to proteins. Proteins with sugar chains attached are called glycoproteins, and the sugar chains are called glycans. There are a few different ways that a sugar chain can be attached to a protein; of relevance here is N-linked glycosylation, where the glycan is covalently bound to a nitrogen (N) atom in an asparagine (N) amino acid in the protein.
Glycans attached to proteins can be quite diverse. This image conveys just a few of the major groups of glycans. (adapted from Berninsone, P.M.):
{kind=link}

Glycans can differ on several dimensions: the length, branching structure, and the particular sugar molecules at each position.
brief review of PrP cell biology
To understand PrP’s glycosylation, it’s useful to have a quick background on PrP’s cell biology. For more details, see my secretory pathway notes or [Harris 2003 (ft)]. Here we go in brief.
Human PrP is 253 amino acids long, but the first 22 are a signal peptide which causes the mRNA/ribosome complex to get moved, mid-translation, to the endoplasmic reticulum (this is ‘cotranslational translocation’). Those 22 amino acids (MANLGCWMLVLFVATWSDLGLC) get cleaved off by signal peptidase and the protein continues to be translated directly into the ER. While it’s still being translated, oligosaccharyl transferase (OST) adds glycan chains at 0, 1 or 2 of 2 possible sites. In general, OST recognizes two amino acid motifs – NXS and NXT – as its sites for attaching glycans. In human PrP these occur at codon 181-183 (NIT) and 197-199 (NFT). But OST’s hit rate is not 100%. Any given molecule of PrP might end up with glycan chains attached at codon 181, codon 197, both, or neither.
The C-terminus of PrP is a 23 amino acid sequence (SMVLFSSPPVILLISFLIFLIVG) which signals for the addition of a GPI anchor. In this process, called glypiation, these 23 amino acids are cleaved off and replaced with GPI, which is itself another kind of sugar chain, which embeds in the membrane, thus anchoring PrP to the ER membrane. Depending who you ask, glypiation may also be called ’glycosylation’, but is not the subject of this post. This post is about the N-linked glycosylation at codons 181 and 197. Once the signal peptide and GPI signal have both been cleaved, the initial 253 amino acid protein is just 208 amino acids.
From its beginnings in the ER, PrP transits through the secretory pathway, acquiring a disulfide bond between its two cysteines (C180 and C214) and undergoing several modifications to its N-linked glycan chains. Secretory pathway proteins with a DXE signal, called a ‘di-acidic’ signal since D is aspartic acid and E is glutamic acid, get exocytosed. PrP has a DYE at codons 144-146, and thus travels from the Golgi to the cell surface in an exocytic vesicle. Once there it remains GPI-anchored to the exoplasmic (outside) surface of the plasma membrane.
how people study PrP glycosylation
Any two molecules of PrP could differ from one another in glycosylation state, in two different ways.
First, as mentioned above, PrP can be glycosylated at N181, N197, both, or neither. That’s four different states. But the way that these can be studied in the lab is by separating them by molecular mass in gel electrophoresis. When this is done, you only see three bands: diglycosylated, monoglycosylated, and unglycosylated. Among the monoglycosylated species of PrP, it’s not possible to distinguish the N181- from the N197-glycosylated molecules.
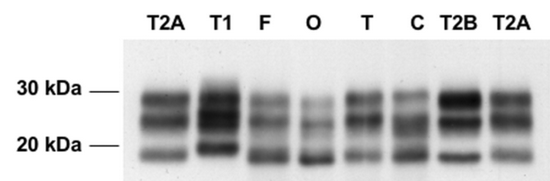
Below: Gel electrophoresis from Levavasseur 2008, Fig 1. PrP (in this case, PrP-res) separates as three bands corresponding to di-, mono- and unglycosylated PrP.
Second, even if two molecules of PrP are both, say, diglycosylated, they might still differ in the types of glycan chains attached (again: length, branching structure, types of sugar molecules included). This is hard to study too. The enzyme PNGase F can be used to cleave the glycan chains off of PrP, and then you can identify the exact glycan chains themselves using mass spectrometry. But you won’t know whether each glycan chain you see came from N181 or N197 *See point 3 in Dr. Kukuskhkin’s comment. Or you can do a two-dimensional gel electrophoresis, where you leave the glycans atatched to PrP and then separate PrP molecules by size along one axis (thus separating di-, mono- and unglycosylated, and probably also separating out different cleavage products) and by isoelectric point on the other axis. Isoelectric point is the pH at which a molecule is neutrally charged, and this differs for many glycan chains, though unlike mass spec, you can’t precisely identify every glycan chain.
Below: 2D gel electrophoresis from Hopf 2011, Fig 4. Proteins (in this image, a bacterial protein, not PrP) are separated by molecular weight on the y axis and isoelectric point on the x axis.

From my reading of the literature, the term ‘glycoform’ seems to be used loosely to refer to either the di/mono/un-glycosylated distinction or to the which-glycan-chain distinction, or to both. I’ll use it loosely here too.
is glycosylation how prion strain information is encoded? (answer: no)
In humans and in other animals, there are different strains of prions, distinguishable pathologically by the specific disease symptoms, incubation times and the brain regions most heavily affected, and distinguishable biochemically by the size of cleavage products and, as we’ll see shortly, the ratio of di-, mono- and un-glycosylated species. These properties are maintained over serial transmission between animals, in the absence of any amino acid sequence difference.
Ever since the notion of infectious proteins was proposed [Griffith 1967, Prusiner 1982], a central mystery has been how proteins could encode different strains. Before the advent of the prion hypothesis, all of the infectious agents that people were used to seeing had nucleic acids, and so differences in DNA or RNA sequence could code for different strains. You might ask: could different strains simply be different glycoforms of PrP?
A considerable body of evidence says the answer is no. The first study to show that glycosylation is not necessary for strain information considered two strains of hamster prions, named hyper (HY) and drowsy (DY) for the different behavioral phenotypes observed in infected hamsters [Bessen 1995]. Bessen discovered that even when PrPSc from each strain was first amplified in a cell-free conversion assay [Kocisko 1994] using purely unglycosylated PrP as a substrate, strain properties were still preserved on transmission to a new set of hamsters. The finding that unglycosylated PrP can preserve strain information has since been replicated in mice with the RML and 301C prion strains [Piro 2009]. No one has yet been able to induce spontaneous PrPC > PrPSc conversion in a cell-free system with wild-type PrP – though several efforts have come very close [Legname 2004, Castilla 2005, Edgeworth 2010, reviewed in Benetti & Legname 2009]. update 2013-06-08: we learned at Prion2013 that Wang 2010 is now accepted as the first successful creation of true de novo prions in a test tube. But we do now also know from studies of yeast prions that conformation alone can be sufficient to encode strain information [Tanaka 2004, Tanaka 2006].
All that is pretty convincing evidence that glycosylation is not necessary for the existence of prion strains. That doesn’t by definition rule out the possibility that glycosylation might still contribute to certain strains, and one recent study has sought to show that passage of prion strains through mice expressing glycosylation mutants does indeed change the strain properties [Cancellotti 2013]. A challenge in interpreting the results of this (and many other in vivo studies, as we’ll see below) is that in order to abolish one or both of PrP’s glycosylation sites, the investigators had to mutate the NXT sequences, thus confounding the effects of glycosylation changes with the effects of amino acid sequence changes.
As an interesting aside, it has recently been shown strain properties can be changed depending upon the cofactors available for prion conversion. Using a version of the protein misfolding cyclic amplification (PMCA) assay [Saborio 2001, Castilla 2005, Deleault 2010], it was shown that the presence or absence of different cofactors can affect the faithfulness of prion strain propagation – specifically, when phosphatidylethanolamine was the only cofactor available, three different prion strains converged into one [Deleault 2012]. This is still consistent with the idea that PrPSc strain information is (or at least can be) encoded in conformation alone, but that cofactors are necessary for the templating of that strain information onto new PrPC molecules. We may eventually learn that glycosylation, similarly, is necessary for the propagation of certain strains, but this hasn’t been shown convincingly yet. But whether or not it’s strictly necessary, there is good evidence that glycosylation does affect the efficiency of prion conversion for various strains, as we’ll see later in this post.
different strains have different glycosylation patterns
Although glycosylation does not appear to be necessary to encode strain information, strains do have clearly different glycosylation patterns.
In humans, different strains of sporadic and acquired Creutzfeldt-Jakob Disease exhibit different relative proportions of un-, mono-, and di-glycosylated species of PrP, and these differences are stable over passage through mice [Collinge 1996]. Similarly reproducible differences in the extent of glycosylation were also observed for several rodent strains of PrPSc [Somerville 1997]. In that latter study, a limited amount of within-strain variation in glycosylation was still observed, and available experimental techniques were not sufficiently precise to uniquely identify all strains based solely on glycosylation state [Somerville 1997]. Still, it was clear that strains have characteristic glycoform ratios. Indeed, the identical glycoform ratio observed in human variant Creutzfeldt-Jakob Disease (vCJD) and bovine spongiform encephalopathy (BSE) upon transmission of each to mice was used as one of several lines of evidence demonstrating that these two strains are one and the same, and that the vCJD epidemic in the U.K. arose from the transmission of BSE or ‘mad cow disease’ to humans [Hill 1997].
In an even more extreme example of characteristic glycoform patterns, it has recently been shown that two human prion diseases – familial Creutzfeldt-Jakob Disease caused by the V180I mutation, and a newly characterized sporadic prion disease dubbed variably protease-sensitive prionopathy (VPSPr) [Zou 2010] – contain no PrPSc molecules glycosylated at N181, and therefore no diglycosylated PrPSc molecules at all [Xiao 2013]. This feature had previously been observed in a familial form of Creutzfeldt-Jakob Disease caused by the T183A mutation, but in that case, the amino acid substitution abolishes the N181 glycosylation site, which requires an NXS or NXT motif. In contrast, VPSPr is a sporadic disease not associated with any amino acid substitution, and cells expressing V180I PrP do produce PrPC glycosylated at N181. Therefore it appears that PrP glycosylated at N181 does exist in both of these diseases, but simply cannot be converted to the particular strain of PrPSc that is present.
PrPSc of different strains may differ not only on the di/mono/unglycosylated dimension, but also in the nature of glycan chains attached. One study has shown a difference in glycan chains between PrPSc in two different human prion diseases: sporadic fatal insomnia (sFI) and the M/M2 subtype of sporadic Creutzfeldt-Jakob Disease (sCJD) [Pan 2001]. These diseases exhibit different phenotypes in the absence of any amino acid substitutions in PrP, and all subjects examined (n=2 for each disease, i.e. n=4 total) were homozygous MM at codon 129. The relative abundance of di-, mono- and un-glycosylated forms was indistinguishable between the two diseases, but sCJD appeared to contain a greater diversity of glycan chains than sFI. 2D gel electrophoresis revealed several dots (corresponding to different isoelectric points) present only in sCJD and not in sFI. These disappeared when the PrPSc was deglycosylated prior to electrophoresis, confirming that it was the glycan chains that caused the different isoelectric points. And no such differences were observed in PrPC isolates, suggesting the observed differences were the product of prion infection and not just differences between the patients’ original sets of glycoforms.
the preferential conversion hypothesis or ‘selection model’
The finding that glycosylation states differ between strains led Collinge to propose two hypotheses: (1) that each PrPSc conformation may have an easier time converting certain glycoforms of PrPC rather than others, and (2) that the selective vulnerability of different brain regions to different prion strains may therefore owe in whole or part to the local abundance of compatible glycosylation species [Collinge 1996].
Hypothesis #1, which I will call the preferential conversion hypothesis and which some authors have called the ‘selection model’, would imply that a given strain (defined by its conformation alone) might efficiently convert, say, monoglycosylated PrP but inefficiently convert diglycosylated PrP. (Or the efficiency might vary by the type of glycan chains attached). This might happen because the glycan chains stabilize a particular structure, or get in the way of conversion, or get in the way of a particular folding conformation, thus raising the energy barrier that has to be crossed to convert from PrPC to a particular conformation of PrPSc.
If this is true, then PrPSc glycoforms should differ from PrPC glycoforms in the same brain. Pan 2001‘s and Xiao 2013‘s results both seem to support this notion, for glycan chain types and for di/mono/unglycosylated PrP molecules respectively. But not all investigators have agreed. Mass spectrometry of glycans isolated from uninfected and infected Syrian hamster brains revealed 52 different glycan chains attached to PrP, every one of which was present in both PrPC and PrPSc [Rudd 1999]. The relative abundance of some glycans differed, with PrPSc enriched for tri- and tetra-antennary and depleted for bi-antennary structures relative to PrPC, but this was speculatively attributed to a global disruption of GnTIII enzyme activity in the course of prion-mediated neurodegeneration and thus considered a consequence of PrPSc infection rather than an intrinsic property of PrPSc.
Perhaps a more testable implication of the preferential conversion model is that prion strains should have trouble infecting mice expressing incompatible glycoforms of PrP. For instance, a strain with a ‘preference’ for diglycosylated PrP should cause mild or no disease in mice that don’t express diglycosylated PrP. As mentioned above, the troubling confounder here is that in order to abolish glycosylation at one of PrP’s N-linked glycosylation sites, you have to mutate either the N or the T in the NXT signal.
That amino acid change matters a lot, because prion propagation efficiency depends on compatibility between the PrPSc amino acid sequence and the target PrPC amino acid sequence [reviewed in Collinge & Clarke 2007]. Prion strains have different ‘species barriers’ which tend to go away after the strain is passaged in the new recipient species or when the recipient animal expresses a transgene of the original species’ PrP sequence [Prusiner 1990] and humans with homozygous codon 129 genotypes are much more vulnerable to sporadic CJD [Palmer 1991] as well as vCJD and have earlier onset in some genetic prion diseases [reviewed in Lukic & Mead 2011]. So it’s to be expected that if you mutate an N or a T in mice to some other amino acid, that might affect vulnerability to prion strains regardless of the resulting glycosylation changes.
Keep that in mind while interpreting the results from mouse studies. Tuzi 2008 created mice with the Ns mutated to Ts in the first, second or both glycosylation sites of PrP (called G1, G2 and G3 mice respectively). The resulting mice were then infected with 79A prions (a low glycosylation strain) or ME7 prions (a medium glycosylation strain). The incubation times of the different strains differed dramatically among the different mouse mutants and versus wild-type mice. Perhaps most convincingly, the G3 mice (with entirely unglycosylated PrP) were capable of sustaining a 79A infection, but they got sick very late in life if ever (only 4 of 21 mice died of prion disease), and they appeared wholly incapable of sustaining an ME7 infection. The G1 mice also could not support ME7 infection, suggesting the first glycosylation site was indispensible for ME7 infection.
Tuzi’s results could all be explained by amino acid sequence changes, or could be explained by glycosylation. Indeed, the story is quite plausible if you think of it in terms of glycosylation: 79A, the ‘low glycosylation’ strain, could infect any of the mutant mice (though with extended incubation times), while ME7, the ‘medium glycosylation’ strain, seemed to require glycosylation at the first site but could do without glycosylation at the second.
The same problem – is it amino acid sequence or glcyosylation that matters – affects interpretation of similar work in cell culture [Salamat 2011] and of the later work showing changes in strain properties after passage through Tuzi’s mice [Cancellotti 2013]. Of note, Salamat discovered that totally unglycosylated mutants of PrP are not properly trafficked to the cell surface, which could account for some of the extended incubation times that Tuzi observed, though it still cannot account for the differences in those effects between the 79A and ME7 prions.
To my knowledge, no study has yet really convincingly separated the effects of glycosylation from amino acid sequence in these mutant PrP models. Therefore these studies cannot be taken as proof of the preferential conversion hypothesis, though overall they do seem to support it.
Rather, the most convincing evidence for the preferential conversion model comes from the fact that prion strains maintain their own characteristic glycoform ratios even in cell-free conversion assays, provided that all the cofactors and a diversity of PrPC glycoforms are available as substrates [Castilla 2008]. In Castilla’s Fig 1 you can see that the respective glycoform ratios of four different prion strains are virtually identical before and after PMCA – and consistently different from each other.
Following Collinge’s original hypotheses, Somerville had proposed two other models to explain the glycoform differences of prion strains, suggesting that strains might differentially affect the glycosylation of PrP at the point of synthesis (similar, but not identical, to Rudd 1999‘s suggestion that enzyme disruption in the disease process led to differences in PrPSc vs. PrPC glycosylation) or that different glycoforms might be differentially deglycosylated by other enzymes during infection by different strains [Somerville 1999]. Neither synthesis nor degradation is occuring in the PMCA assay, so preferential conversion seems to be the only model to explain the maintenance of glycoform ratios. In interpreting his own data, Castilla concludes that the selection model must be correct [Castilla 2008].
selective vulnerability of different brain regions
If strains do exhibit a ‘preference’ for converting different glycoforms of PrP, that could go a long ways towards explaining why strains preferentially affect different brain regions, a phenomenon called strain-specific neurotropism.
Total PrPC levels differ across the brain, being highest in the thalamus [DeArmond 1999]. But that’s true regardless of what strain an animal is infected with, so differences in host PrP expression could explain at most one strain’s neurotropism (brain region preference). Some other brain region difference is needed to explain the neurotropic differences between strains. And indeed, PrPC glycosylation does differ across brain regions. 2D gel electrophoresis has been used to separate PrPC molecules both by size and by isoelectric point, showing differences in the glycan chains of diglycosylated PrPC across several different mouse brain regions [DeArmond 1997]. Later studies using gel electrophoresis or antibodies with differential affinity for di-, mono- or un-glycosylated species of PrP were also able to show differences in the prevalence of these species across different brain tissues both in mice [Somerville 1999 (ft), Beringue 2003] and in humans [Kuczius 2007]. All of these findings for PrP specifically are consistent with the well-established knowledge that, for glycoproteins generally, glycosylation patterns are tissue-specific [Rademacher 1988].
Together these results suggest sufficient heterogeneity in PrP glycoforms across different brain regions to potentially account, at least in part, for selective vulnerability.
But to my knowledge no one has really been able to show this experimentally. To do so would seem to require somehow changing the glycoform ratios (and not changing anything else) in different brain regions and then showing that this alters which brain region is affected by which strain. That’s a tall order, especially when we don’t have the tools to even convincingly separate glycosylation changes from amino acid changes in the whole organism. *See point 5 below.
On the other hand, in a quick thought experiment, I find it hard to talk myself into the idea that glycosylation doesn’t matter for neurotropism. As discussed above, it’s clear that the relative ratios of glycoforms of PrPSc collected from brains of humans or other animals do differ between strains, sometimes dramatically so. That’s consistent with the idea that strains have preferences for certain glycoforms, and that those preferences drive them to accumulate in different brain regions where their preferred glycoforms are abundant. One alternate interpretation of this data would be that different strains affect different brain regions for some other, unobserved, non-glycosylation-related reason, and that then because they are active in different brain regions, they end up converting the glycoforms that are locally available. That alternate interpretation is hard to reconcile with the existence of glycosylation preferences in vitro [Castilla 2008].
But that’s not proof, and in any event, glycosylation could contribute to strain-specific neurotropism without explaining all of it. For instance, since it’s clear that cofactors matter too [Deleault 2012], the availability of different cofactors across brain regions could also contribute to explaining it.
All this is assuming an implicit causal link: that the amount of PrPSc produced in each brain region determines the extent of pathology there. The couple of studies I was able to find do suggest that PrPSc levels and pathology are pretty strongly correlated across brain regions, both in CJD [Parchi 1996] and FFI [Cortelli 1997]. Interpretation of this is complicated by the fact that when we say “PrPSc“, what is usually being measured is actually proteinase K resistant PrP (PrP-res), or PrP plaque staining, both of which appear to be just proxies for disease and not direct measures of what is toxic, and FFI doesn’t produce much PrP-res anyway [Jackson 2009].
summary of the literature
Based on my above read-through of the literature, the following points seem very well-supported by available evidence:
- Glycosylation is not necessary for encoding strain information in prions.
- Prion strains exhibit reproducible glycoform ratios which are faithfully maintained during propagation both in vitro and in vivo.
- Prion strains have different ‘preferences’ for converting different PrP glycoforms.
- Ratios of PrPC glycoforms differ across brain regions.
All this suggests a role for glycobiology in explaining strain-specific neurotropism, but this has never been experimentally demonstrated.
is glycosylation a potential therapeutic target in prion disease?
At first glance, based on the above evidence, it would seem that targeting glycosylation could be a therapeutic strategy, but not an ideal one. For one, since glycosylation ‘preferences’ differ between prion strains, glycosylation-based strategies seem likely to have strain-specific efficacy, which would limit their usefulness. Second, with a few exceptions (VPSPr, V180I CJD [Xiao 2013] and ME7 in G3 mice [Tuzi 2008]) the glycoform ‘preferences’ of prion strains do not appear to be absolute. That is, a strain may ‘prefer’ to convert diglycosylated PrP but still can convert un- or monoglycosylated PrP. That suggests that changing the glycosylation state of PrPC might delay prion disease a bit, but not forever.
Two studies that examined prion replication in the presence of compounds that alter glycosylation seem to support these conjectures [Browning 2011, Oelschlegel & Weissmann 2013]. These studies were not exactly therapeutic-oriented, but rather, used glycosylation-altering compounds as tools to probe the hypothesis that each prion strain is actually a quasi-species of several substrains, which can be selected for or against by different drugs. In addition to their biochemical and phenotypic properties, prion strains can now be distinguished by their relative ability to infect different cell lines, and their relative resistance to different strain-specific drugs such as curcumin and cpd-B, in a ‘cell panel assay’ [Mahal 2007]. Scrapie-infected cells were treated with a variety of glycosylation-modifying drugs and other small molecules, including kifunensine (kifu), castanospermine (CST) and swainsonine (swa). In support of the ‘quasi-species’ or ‘many substrains’ hypothesis, some strains of prions were initially sensitive to some drugs, but could develop drug resistance or drug ‘dependence’ (increased efficiency of propagation in presence of the drug) after being cultured in the presence of the drug [Browning 2011, Oelschlegel & Weissmann 2013].
The strain specificity of these compounds, and the related fact that they may merely select for resistant substrains like quinacrine does [Ghaemmagami & Ahn 2009], indeed seems to suggest they wouldn’t be ideal therapeutics, though these studies did not examine in vivo efficacy.
But another property of glycosylation-altering drugs is their ability to in some cases increase the degradation of glycoproteins, reducing the total amount of the protein produced. That could fit into the therapeutic strategy of depleting PrP, a possibility which I’ll consider briefly here.
My interest in learning about PrP glycosylation and writing this post was triggered by a conversation I had with glycobiologist Nikolay Kukushkin. He pointed out that alpha glucosidase inhibitors, a class which includes several approved drugs (and the experimental compound CST), can not only alter glycosylation but also reduce the total amount of some glycoproteins produced. As one example we discussed a drug called n-butyl-deoxynorijimycin (NB-DNJ; commonly known as Miglustat and commercially known as Zavesca; shown below), which is prescribed for Gaucher disease and Niemann-Pick Type C.

This drug’s mechanism of action is fascinating. Its therapeutic value in the storage diseases Gaucher and Niemann-Pick is due to its inhibition of ceramide glucosyltranferase, thus reducing the amount of certain glycolipids produced and lessening the overall burden of storage material. This approach is called ‘substrate reduction therapy’. However, many of the glycan-processing enzymes have similar enough binding sites that one drug may inhibit several of them. It so happens [sic, see point 6] that NB-DNJ also inhibits alpha glucosidases [see for instance Fischer 1995, Tian 2004], enzymes which normally trim the glycan chains of a freshly synthesized, immature glycoprotein in the ER, allowing that glycoprotein to interact with folding chaperones calnexin/calreticulin. The protein will fold in the ER with their help, and then later in the Golgi, it will have its glycan chains further extended and modified by other enzymes. When glycans are not trimmed (due to NB-DNJ), the glycoprotein cannot interact with calnexin/calreticulin, and remains in its initial unfolded state with immature glycan chains attached. That state doesn’t last forever: our cells have redundant mechanisms for trimming the glycan chains later in the Golgi, and so eventually the glycan chains will be extended and modified like always. But this redundancy isn’t perfect, so NB-DNJ does result in changes to the final glycosylation state of some proteins, and more importantly, the added time that the glycoprotein spends in an immature and unfolded state in the ER gives it more chances to be degraded (via ERAD) before it ever passages through the Golgi. This means that the total amount of some glycoproteins is reduced by NB-DNJ.
NB-DNJ’s inhibition of alpha glucosidases leads to totally non-specific downstream effects – it affects all glycoproteins – and raises the interesting question of how it would affect PrP. I did PubMed and Google Scholar searches for ‘prion n-butyl-deoxynorijimycin’ and similar search terms and could not find any studies that have examined this question. NB-DNJ/Miglustat is also not included in MSDI’s Spectrum Collection recently used in the Ghaemmagami lab’s ELISA assay for PrP-res inhibitors [Poncet-Montagne 2011] nor in MSDI’s US Drug Collection used in the Lasmezas lab’s FRET assay for PrP reducers [Karapetyan & Sferrazza 2013], so there doesn’t seem to be any available high-throughput screening evidence on its effects either.
We can make a reasonable conjecture that any glycosylation-altering effects of NB-DNJ would result in strain-specific inhibitory effects, as seen for CST [Browning 2011]. But the experiments in these studies were run on cell lines expressing wild-type PrP. It is interesting to consider the possibility that in mutant cell lines, alpha glucosidase inhibitors might increase the degradation of the mutant protein.
That bit of speculation is based on a study looked at the metabolism and secretion of PrP in cell lines transfected with DNA expressing either of the D178N mutants: D178N cis 129M FFI and D178N cis 129V CJD [Petersen 1996]. In both cases, Petersen found that the unglycosylated form of the mutant (but not wild-type) PrP was almost completely degraded before reaching the cell surface. At the time of synthesis in the ER, unglycosylated PrP appeared to account for ~20% of total PrP in both the mutant and wild-type varieties, but by the time it reached the cell surface, unglycosylated D178N 129M PrP was undetectable, and unglycosylated D178N 129V PrP had dropped to 0.6% of total PrP. Petersen then looked at postmortem brains of FFI patients, which confirmed that the same phenomenon is present in vivo, albeit to a somewhat lesser extent than in cell culture. Unglycosylated mutant PrP was under-represented compared to unglycosylated PrP from the patient’s wild-type allele, suggesting that it might be preferentially degraded.
Petersen’s results can be taken to suggest that in the absence of glycan chains, the mutant PrP misfolds in some way that triggers a cellular response to degrade it. *Or not – see point 7. Could a similar effect be achieved through alpha glucosidase inhibitors, promoting early degradation in the ER? It’s a risky proposition: by promoting misfolding of some sort, the drugs might accelerate prion disease. Indeed, tunicamycin, a toxic, non-drug experimental compound which blocks glycosylation altogether is used as a positive control for ER stress [for instance in Moreno 2012] because it causes such a dramatic accumulation of misfolded proteins. It’s also not clear if ER-associated degradation could be achieved: Petersen found evidence that the degradation of unglycosylated mutant PrP in some late endosomal/lysosomal compartment, rather than ERAD. More recent work has agreed that mutant PrP degradation appears to take place in acidic compartments past the Golgi, rather than in the ER [Ashok & Hedge 2009].
To my knowledge, no research has yet examined whether alpha glucosidase inhibitors can promote early degradation of mutant PrP before it reaches the cell surface. A first step would be to test the effects of alpha glucosidase inhibitors in cell lines expressing mutant PrP and check for reduced expression of the mutant allele. If expression indeed appeared to be reduced, effects in vivo could be determined in knock-in mice. If it happened to have therapeutic value, that would be exciting -NB-DNJ has good blood-brain barrier penetrance [Patterson 2007] and is an approved drug with an apparently decent safety record [Ficicioglu 2008].
conclusions
N-linked glycosylation turns out to be a fascinating part of prion biology. Different strains of prions exhibit different characteristic glycoform patterns, a phenomenon which has now been shown, fairly conclusively, to be due to the ‘preference’ of different strains for converting certain glycoforms of PrPC. This ‘preference’ might even contribute to explaining why strains of prions affect different brain regions more dramatically.
Glycosylation is not an ideal therapeutic target in prion diseases, because its effects would be likely to be strain-dependent, and because for the most part the ‘preferences’ of different strains are not absolute, so even at best, altering glycosylation could only slow, and not stop, prion disease. Still, it would be interesting to know whether alpha glucosidase inhibitors, a class of drugs which can alter glycosylation and/or increase degradation of some proteins, could reduce expression of mutant PrP. These have apparently never been tested for their effects on genetic mutants of PrP.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.